# import all needed R packages
library(ChAMP)
library(ChAMPdata)
library(ggplot2)
library(stringr)
library(ggpubr)
Lade nötiges Paket: minfi
Lade nötiges Paket: BiocGenerics
Lade nötiges Paket: generics
Attache Paket: ‘generics’
Die folgenden Objekte sind maskiert von ‘package:base’:
as.difftime, as.factor, as.ordered, intersect, is.element, setdiff,
setequal, union
Attache Paket: ‘BiocGenerics’
Die folgenden Objekte sind maskiert von ‘package:stats’:
IQR, mad, sd, var, xtabs
Die folgenden Objekte sind maskiert von ‘package:base’:
anyDuplicated, aperm, append, as.data.frame, basename, cbind,
colnames, dirname, do.call, duplicated, eval, evalq, Filter, Find,
get, grep, grepl, is.unsorted, lapply, Map, mapply, match, mget,
order, paste, pmax, pmax.int, pmin, pmin.int, Position, rank,
rbind, Reduce, rownames, sapply, saveRDS, table, tapply, unique,
unsplit, which.max, which.min
Lade nötiges Paket: GenomicRanges
Lade nötiges Paket: stats4
Lade nötiges Paket: S4Vectors
Attache Paket: ‘S4Vectors’
Das folgende Objekt ist maskiert ‘package:utils’:
findMatches
Die folgenden Objekte sind maskiert von ‘package:base’:
expand.grid, I, unname
Lade nötiges Paket: IRanges
Lade nötiges Paket: GenomeInfoDb
Lade nötiges Paket: SummarizedExperiment
Lade nötiges Paket: MatrixGenerics
Lade nötiges Paket: matrixStats
Attache Paket: ‘MatrixGenerics’
Die folgenden Objekte sind maskiert von ‘package:matrixStats’:
colAlls, colAnyNAs, colAnys, colAvgsPerRowSet, colCollapse,
colCounts, colCummaxs, colCummins, colCumprods, colCumsums,
colDiffs, colIQRDiffs, colIQRs, colLogSumExps, colMadDiffs,
colMads, colMaxs, colMeans2, colMedians, colMins, colOrderStats,
colProds, colQuantiles, colRanges, colRanks, colSdDiffs, colSds,
colSums2, colTabulates, colVarDiffs, colVars, colWeightedMads,
colWeightedMeans, colWeightedMedians, colWeightedSds,
colWeightedVars, rowAlls, rowAnyNAs, rowAnys, rowAvgsPerColSet,
rowCollapse, rowCounts, rowCummaxs, rowCummins, rowCumprods,
rowCumsums, rowDiffs, rowIQRDiffs, rowIQRs, rowLogSumExps,
rowMadDiffs, rowMads, rowMaxs, rowMeans2, rowMedians, rowMins,
rowOrderStats, rowProds, rowQuantiles, rowRanges, rowRanks,
rowSdDiffs, rowSds, rowSums2, rowTabulates, rowVarDiffs, rowVars,
rowWeightedMads, rowWeightedMeans, rowWeightedMedians,
rowWeightedSds, rowWeightedVars
Lade nötiges Paket: Biobase
Welcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages 'citation("pkgname")'.
Attache Paket: ‘Biobase’
Das folgende Objekt ist maskiert ‘package:MatrixGenerics’:
rowMedians
Die folgenden Objekte sind maskiert von ‘package:matrixStats’:
anyMissing, rowMedians
Lade nötiges Paket: Biostrings
Lade nötiges Paket: XVector
Attache Paket: ‘Biostrings’
Das folgende Objekt ist maskiert ‘package:base’:
strsplit
Lade nötiges Paket: bumphunter
Lade nötiges Paket: foreach
Lade nötiges Paket: iterators
Lade nötiges Paket: parallel
Lade nötiges Paket: locfit
locfit 1.5-9.12 2025-03-05
Setting options('download.file.method.GEOquery'='auto')
Setting options('GEOquery.inmemory.gpl'=FALSE)
Lade nötiges Paket: ChAMPdata
Lade nötiges Paket: DMRcate
Lade nötiges Paket: Illumina450ProbeVariants.db
Lade nötiges Paket: IlluminaHumanMethylationEPICmanifest
Lade nötiges Paket: DT
Lade nötiges Paket: RPMM
Lade nötiges Paket: cluster
Keine Methoden in Paket 'RSQLite' gefunden für Anforderung: 'dbListFields' beim Laden von 'lumi'
Warning message:
"vorhergehender Import 'plyr::mutate' durch 'plotly::mutate' während des Ladens von 'ChAMP' ersetzt"
Warning message:
"vorhergehender Import 'plyr::rename' durch 'plotly::rename' während des Ladens von 'ChAMP' ersetzt"
Warning message:
"vorhergehender Import 'plyr::arrange' durch 'plotly::arrange' während des Ladens von 'ChAMP' ersetzt"
Warning message:
"vorhergehender Import 'plyr::summarise' durch 'plotly::summarise' während des Ladens von 'ChAMP' ersetzt"
Warning message:
"vorhergehender Import 'plotly::subplot' durch 'Hmisc::subplot' während des Ladens von 'ChAMP' ersetzt"
Warning message:
"vorhergehender Import 'plyr::summarize' durch 'Hmisc::summarize' während des Ladens von 'ChAMP' ersetzt"
Warning message:
"vorhergehender Import 'plyr::is.discrete' durch 'Hmisc::is.discrete' während des Ladens von 'ChAMP' ersetzt"
Warning message:
"vorhergehender Import 'plotly::last_plot' durch 'ggplot2::last_plot' während des Ladens von 'ChAMP' ersetzt"
Warning message:
"vorhergehender Import 'globaltest::model.matrix' durch 'stats::model.matrix' während des Ladens von 'ChAMP' ersetzt"
Warning message:
"vorhergehender Import 'globaltest::p.adjust' durch 'stats::p.adjust' während des Ladens von 'ChAMP' ersetzt"
>> Package version 2.29.1 loaded <<
___ _ _ __ __ ___
/ __| |_ /_\ | \/ | _ \
| (__| ' \ / _ \| |\/| | _/
\___|_||_/_/ \_\_| |_|_|
------------------------------
If you have any question or suggestion about ChAMP, please email to champ450k@gmail.com.
Thank you for citating ChAMP:
Yuan Tian, Tiffany J Morris, Amy P Webster, Zhen Yang, Stephan Beck, Andrew Feber, Andrew E Teschendorff; ChAMP: updated methylation analysis pipeline for Illumina BeadChips, Bioinformatics, btx513, https://doi.org/10.1093/bioinformatics/btx513 REQUIRE ChAMPdata >= 2.23.1
mystery_1 <- "../../example_datasets/epicv2/mystery_data_1"
mystery_2 <- "../../example_datasets/epicv2/mystery_data_2"
Analyse mystery_data_1
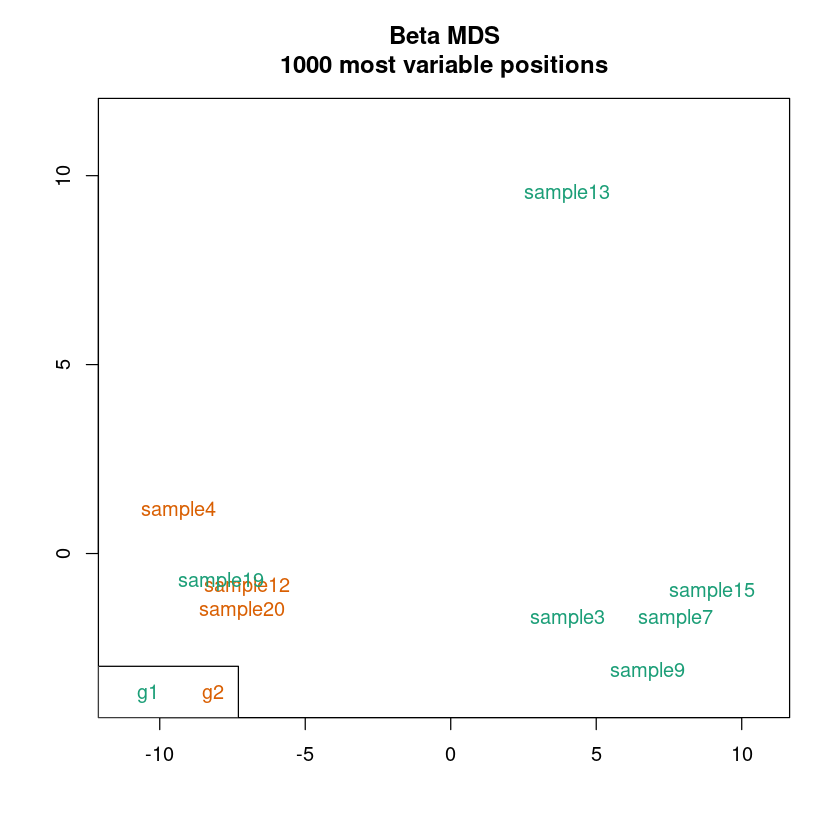
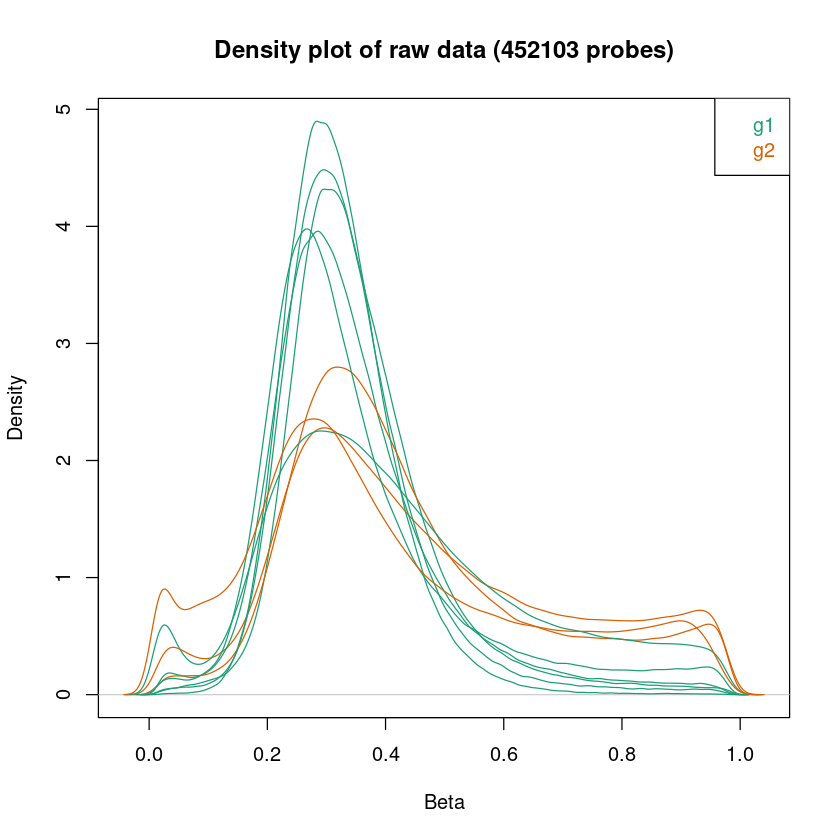
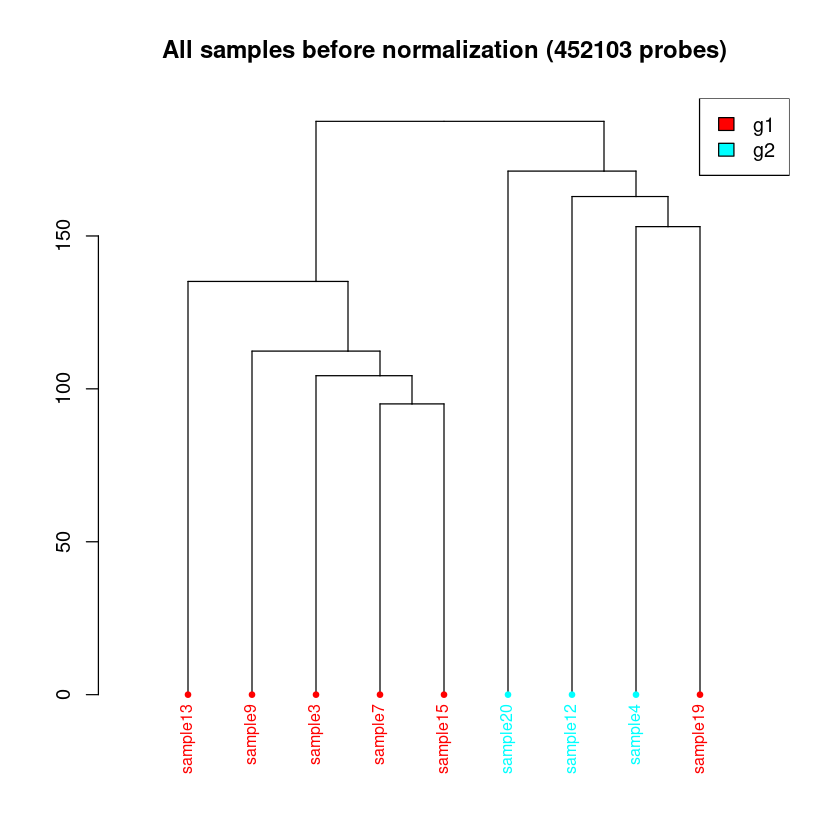
Laden Sie den Datensatz im Verzeichnis mystery_data_1 und schauen Sie sich den Output von champ.laod die QC Plots an:
- Was fällt Ihnen am Output von champ.load auf?
- Wozu führt die Änderung des detPcut Parameters in champ.load?
- Warum wurde der Parameter verändert?
- Was fällt Ihnen an den Plots auf?
- Worauf könnte das zu sehende hindeuten?
- Wie könnten Sie damit umgehen?
# all parameters except one are default
myData_1 <- champ.load(directory = fragmented,
method="ChAMP",
methValue="B",
autoimpute=TRUE,
filterDetP=TRUE,
ProbeCutoff=0,
SampleCutoff=0.1,
detPcut=0.9, # This is the only parameter which was changed compared to the default settings of champ.load
filterBeads=TRUE,
beadCutoff=0.05,
filterNoCG=TRUE,
filterSNPs=TRUE,
population=NULL,
filterMultiHit=TRUE,
filterXY=TRUE,
force=FALSE,
arraytype="EPICv2")
[===========================]
[<<<< ChAMP.LOAD START >>>>>]
-----------------------------
[ Loading Data with ChAMP Method ]
----------------------------------
Note that ChAMP method will NOT return rgSet or mset, they object defined by minfi. Which means, if you use ChAMP method to load data, you can not use SWAN or FunctionNormliazation method in champ.norm() (you can use BMIQ or PBC still). But All other function should not be influenced.
[===========================]
[<<<< ChAMP.IMPORT START >>>>>]
-----------------------------
[ Section 1: Read PD Files Start ]
CSV Directory: ../../example_datasets/epicv2/mystery_data_1/samplesheet.csv
Find CSV Success
Reading CSV File
Replace Sentrix_Position into Array
Replace Sentrix_ID into Slide
[ Section 1: Read PD file Done ]
[ Section 2: Read IDAT files Start ]
Loading:../../example_datasets/epicv2/mystery_data_1/data_directory/111111111111/111111111111_R05C01_Grn.idat ---- (1/20)
Loading:../../example_datasets/epicv2/mystery_data_1/data_directory/111111111111/111111111111_R06C01_Grn.idat ---- (2/20)
Loading:../../example_datasets/epicv2/mystery_data_1/data_directory/111111111111/111111111111_R07C01_Grn.idat ---- (3/20)
Loading:../../example_datasets/epicv2/mystery_data_1/data_directory/111111111111/111111111111_R08C01_Grn.idat ---- (4/20)
Loading:../../example_datasets/epicv2/mystery_data_1/data_directory/222222222222/222222222222_R01C01_Grn.idat ---- (5/20)
Loading:../../example_datasets/epicv2/mystery_data_1/data_directory/222222222222/222222222222_R02C01_Grn.idat ---- (6/20)
Loading:../../example_datasets/epicv2/mystery_data_1/data_directory/222222222222/222222222222_R03C01_Grn.idat ---- (7/20)
Loading:../../example_datasets/epicv2/mystery_data_1/data_directory/222222222222/222222222222_R04C01_Grn.idat ---- (8/20)
Loading:../../example_datasets/epicv2/mystery_data_1/data_directory/222222222222/222222222222_R05C01_Grn.idat ---- (9/20)
Loading:../../example_datasets/epicv2/mystery_data_1/data_directory/222222222222/222222222222_R06C01_Grn.idat ---- (10/20)
Loading:../../example_datasets/epicv2/mystery_data_1/data_directory/222222222222/222222222222_R07C01_Grn.idat ---- (11/20)
Loading:../../example_datasets/epicv2/mystery_data_1/data_directory/222222222222/222222222222_R08C01_Grn.idat ---- (12/20)
Loading:../../example_datasets/epicv2/mystery_data_1/data_directory/333333333333/333333333333_R01C01_Grn.idat ---- (13/20)
Loading:../../example_datasets/epicv2/mystery_data_1/data_directory/333333333333/333333333333_R02C01_Grn.idat ---- (14/20)
Loading:../../example_datasets/epicv2/mystery_data_1/data_directory/333333333333/333333333333_R03C01_Grn.idat ---- (15/20)
Loading:../../example_datasets/epicv2/mystery_data_1/data_directory/333333333333/333333333333_R04C01_Grn.idat ---- (16/20)
Loading:../../example_datasets/epicv2/mystery_data_1/data_directory/333333333333/333333333333_R05C01_Grn.idat ---- (17/20)
Loading:../../example_datasets/epicv2/mystery_data_1/data_directory/333333333333/333333333333_R06C01_Grn.idat ---- (18/20)
Loading:../../example_datasets/epicv2/mystery_data_1/data_directory/333333333333/333333333333_R07C01_Grn.idat ---- (19/20)
Loading:../../example_datasets/epicv2/mystery_data_1/data_directory/333333333333/333333333333_R08C01_Grn.idat ---- (20/20)
Loading:../../example_datasets/epicv2/mystery_data_1/data_directory/111111111111/111111111111_R05C01_Red.idat ---- (1/20)
Loading:../../example_datasets/epicv2/mystery_data_1/data_directory/111111111111/111111111111_R06C01_Red.idat ---- (2/20)
Loading:../../example_datasets/epicv2/mystery_data_1/data_directory/111111111111/111111111111_R07C01_Red.idat ---- (3/20)
Loading:../../example_datasets/epicv2/mystery_data_1/data_directory/111111111111/111111111111_R08C01_Red.idat ---- (4/20)
Loading:../../example_datasets/epicv2/mystery_data_1/data_directory/222222222222/222222222222_R01C01_Red.idat ---- (5/20)
Loading:../../example_datasets/epicv2/mystery_data_1/data_directory/222222222222/222222222222_R02C01_Red.idat ---- (6/20)
Loading:../../example_datasets/epicv2/mystery_data_1/data_directory/222222222222/222222222222_R03C01_Red.idat ---- (7/20)
Loading:../../example_datasets/epicv2/mystery_data_1/data_directory/222222222222/222222222222_R04C01_Red.idat ---- (8/20)
Loading:../../example_datasets/epicv2/mystery_data_1/data_directory/222222222222/222222222222_R05C01_Red.idat ---- (9/20)
Loading:../../example_datasets/epicv2/mystery_data_1/data_directory/222222222222/222222222222_R06C01_Red.idat ---- (10/20)
Loading:../../example_datasets/epicv2/mystery_data_1/data_directory/222222222222/222222222222_R07C01_Red.idat ---- (11/20)
Loading:../../example_datasets/epicv2/mystery_data_1/data_directory/222222222222/222222222222_R08C01_Red.idat ---- (12/20)
Loading:../../example_datasets/epicv2/mystery_data_1/data_directory/333333333333/333333333333_R01C01_Red.idat ---- (13/20)
Loading:../../example_datasets/epicv2/mystery_data_1/data_directory/333333333333/333333333333_R02C01_Red.idat ---- (14/20)
Loading:../../example_datasets/epicv2/mystery_data_1/data_directory/333333333333/333333333333_R03C01_Red.idat ---- (15/20)
Loading:../../example_datasets/epicv2/mystery_data_1/data_directory/333333333333/333333333333_R04C01_Red.idat ---- (16/20)
Loading:../../example_datasets/epicv2/mystery_data_1/data_directory/333333333333/333333333333_R05C01_Red.idat ---- (17/20)
Loading:../../example_datasets/epicv2/mystery_data_1/data_directory/333333333333/333333333333_R06C01_Red.idat ---- (18/20)
Loading:../../example_datasets/epicv2/mystery_data_1/data_directory/333333333333/333333333333_R07C01_Red.idat ---- (19/20)
Loading:../../example_datasets/epicv2/mystery_data_1/data_directory/333333333333/333333333333_R08C01_Red.idat ---- (20/20)
Extract Mean value for Green and Red Channel Success
Your Red Green Channel contains 1105209 probes.
[ Section 2: Read IDAT Files Done ]
[ Section 3: Use Annotation Start ]
Reading EPICv2 Annotation >>
!!! Important, since version 2.29.1, ChAMP set default `EPIC` arraytype as EPIC version 2.
You can set 'EPIC' or 'EPICv2' to use version 2 EPIC annotation
If you want to use the old version (v1), please specify arraytype parameter as `EPICv1`.
For 450K array, still use `450K`
Fetching NEGATIVE ControlProbe.
Totally, there are 411 control probes in Annotation.
Your data set contains 411 control probes.
Generating Meth and UnMeth Matrix
Extracting Meth Matrix...
Totally there are 937055 Meth probes in EPICv2 Annotation.
Your data set contains 937055 Meth probes.
Extracting UnMeth Matrix...
Totally there are 937055 UnMeth probes in EPICv2 Annotation.
Your data set contains 937055 UnMeth probes.
Generating beta Matrix
Generating M Matrix
Generating intensity Matrix
Calculating Detect P value
Counting Beads
[ Section 3: Use Annotation Done ]
[<<<<< ChAMP.IMPORT END >>>>>>]
[===========================]
[You may want to process champ.filter() next.]
[===========================]
[<<<< ChAMP.FILTER START >>>>>]
-----------------------------
In New version ChAMP, champ.filter() function has been set to do filtering on the result of champ.import(). You can use champ.import() + champ.filter() to do Data Loading, or set "method" parameter in champ.load() as "ChAMP" to get the same effect.
This function is provided for user need to do filtering on some beta (or M) matrix, which contained most filtering system in champ.load except beadcount. User need to input beta matrix, pd file themselves. If you want to do filterintg on detP matrix and Bead Count, you also need to input a detected P matrix and Bead Count information.
Note that if you want to filter more data matrix, say beta, M, intensity... please make sure they have exactly the same rownames and colnames.
[ Section 1: Check Input Start ]
You have inputed beta,intensity for Analysis.
pd file provided, checking if it's in accord with Data Matrix...
pd file check success.
Parameter filterDetP is TRUE, checking if detP in accord with Data Matrix...
detP check success.
Parameter filterBeads is TRUE, checking if beadcount in accord with Data Matrix...
beadcount check success.
parameter autoimpute is TRUE. Checking if the conditions are fulfilled...
!!! ProbeCutoff is 0, which means you have no needs to do imputation. autoimpute has been reset FALSE.
Checking Finished :filterDetP,filterBeads,filterMultiHit,filterSNPs,filterNoCG,filterXY would be done on beta,intensity.
You also provided :detP,beadcount .
[ Section 1: Check Input Done ]
[ Section 2: Filtering Start >>
Filtering Detect P value Start
The fraction of failed positions per sample
You may need to delete samples with high proportion of failed probes:
Failed CpG Fraction.
sample1 0.10990604
sample2 0.12215718
sample3 0.08568867
sample4 0.05344083
sample5 0.10018195
sample6 0.13114598
sample7 0.09822582
sample8 0.14327761
sample9 0.08778353
sample10 0.12545261
sample11 0.10311028
sample12 0.05736163
sample13 0.09452807
sample14 0.12113803
sample15 0.09212480
sample16 0.11706463
sample17 0.11174371
sample18 0.10414223
sample19 0.05110693
sample20 0.04452780
The detSamplecut parameter is : 0.1
Samples : sample1,sample2,sample5,sample6,sample8,sample10,sample11,sample14,sample16,sample17,sample18 will be deleted.
There are 9 samples remained for analysis.
Filtering probes with a detection p-value above 0.9.
Removing 440434 probes.
If a large number of probes have been removed, ChAMP suggests you to identify potentially bad samples
Filtering BeadCount Start
Filtering probes with a beadcount <3 in at least 5% of samples.
Removing 12688 probes
Filtering NoCG Start
Only Keep CpGs, removing 1292 probes from the analysis.
Filtering SNPs Start
!!! Important, since version 2.29.1, ChAMP set default `EPIC` arraytype as EPIC version 2.
You can set 'EPIC' or 'EPICv2' to use version 2 EPIC annotation
If you want to use the old version (v1), please specify arraytype parameter as `EPICv1`.
For 450K array, still use `450K`
Using general mask options
Removing 20000 probes from the analysis.
Filtering MultiHit Start
Filtering probes that align to multiple locations as identified in Nordlund et al
Removing 0 probes from the analysis.
Filtering XY Start
Filtering probes located on X,Y chromosome, removing 10538 probes from the analysis.
Updating PD file
Fixing Outliers Start
Replacing all value smaller/equal to 0 with smallest positive value.
Replacing all value greater/equal to 1 with largest value below 1..
[ Section 2: Filtering Done ]
All filterings are Done, now you have 452103 probes and 9 samples.
[<<<<< ChAMP.FILTER END >>>>>>]
[===========================]
[You may want to process champ.QC() next.]
[<<<<< ChAMP.LOAD END >>>>>>]
[===========================]
[You may want to process champ.QC() next.]
champ.QC(beta = myData_1$beta,
pheno=myData_1$pd$Sample_Group,
resultsDir="./CHAMP_QCimages/")
[===========================]
[<<<<< ChAMP.QC START >>>>>>]
-----------------------------
champ.QC Results will be saved in ./CHAMP_QCimages/
[QC plots will be proceed with 452103 probes and 9 samples.]
<< Prepare Data Over. >>
<< plot mdsPlot Done. >>
<< Plot densityPlot Done. >>
< Dendrogram Plot Feature Selection Method >: No Selection, directly use all CpGs to calculate distance matrix.
<< Plot dendrogram Done. >>
[<<<<<< ChAMP.QC END >>>>>>>]
[===========================]
[You may want to process champ.norm() next.]
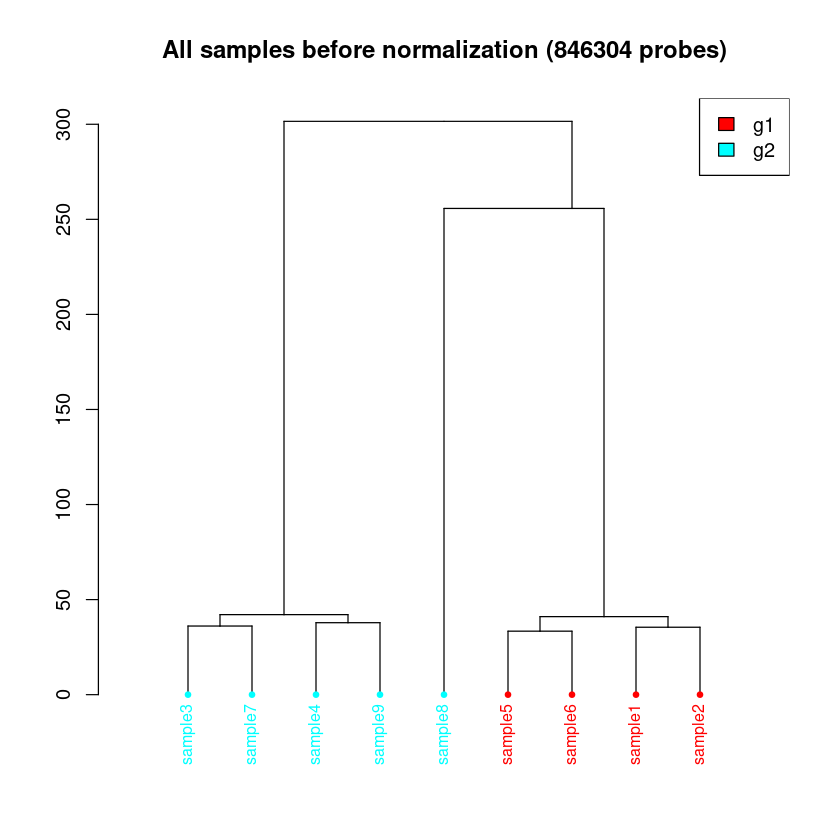
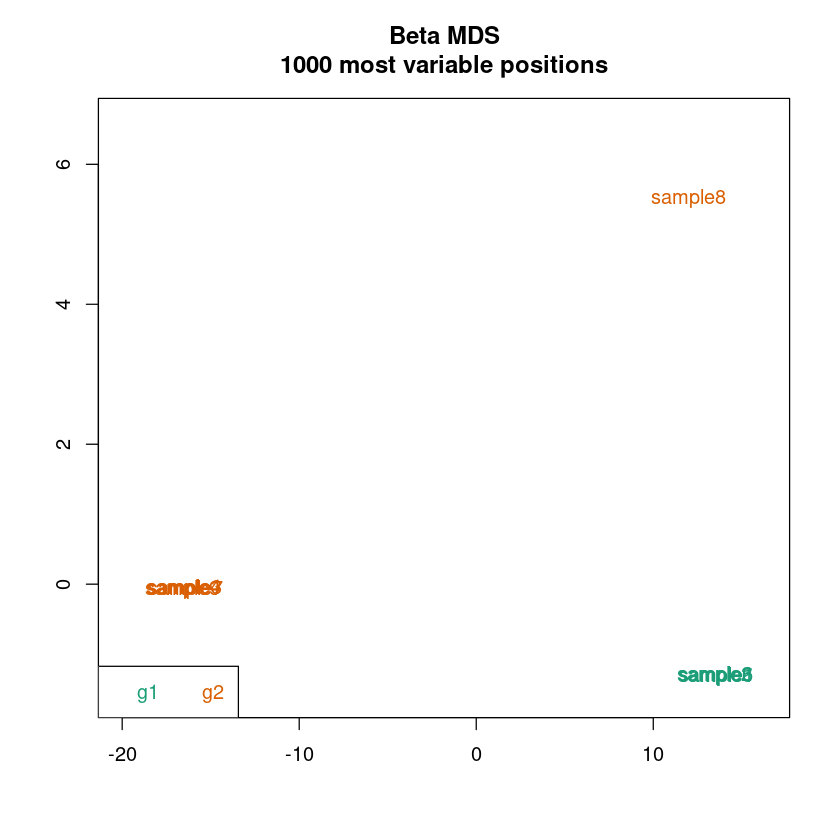
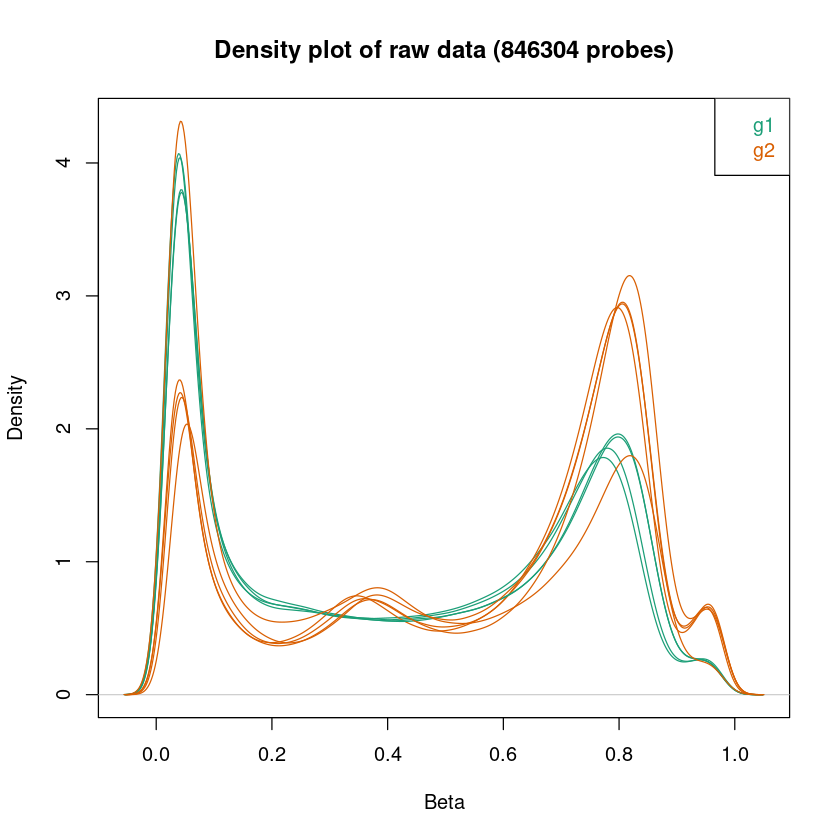
Analyse mystery_data_2
Laden Sie den Datensatz im Verzeichnis mystery_data_2 und schauen Sie sich die QC Plots an:
- Was fällt Ihnen an den Plots auf?
- Worauf könnte das zu sehende hindeuten?
- Wie könnten Sie damit umgehen?
# parameters are all default
myData_2 <- champ.load(directory = intra_group_outlier,
method="ChAMP",
methValue="B",
autoimpute=TRUE,
filterDetP=TRUE,
ProbeCutoff=0,
SampleCutoff=0.1,
detPcut=0.01,
filterBeads=TRUE,
beadCutoff=0.05,
filterNoCG=TRUE,
filterSNPs=TRUE,
population=NULL,
filterMultiHit=TRUE,
filterXY=TRUE,
force=FALSE,
arraytype="EPICv2")
[===========================]
[<<<< ChAMP.LOAD START >>>>>]
-----------------------------
[ Loading Data with ChAMP Method ]
----------------------------------
Note that ChAMP method will NOT return rgSet or mset, they object defined by minfi. Which means, if you use ChAMP method to load data, you can not use SWAN or FunctionNormliazation method in champ.norm() (you can use BMIQ or PBC still). But All other function should not be influenced.
[===========================]
[<<<< ChAMP.IMPORT START >>>>>]
-----------------------------
[ Section 1: Read PD Files Start ]
CSV Directory: ../../example_datasets/epicv2/mystery_data_2/samplesheet.csv
Find CSV Success
Reading CSV File
Replace Sentrix_Position into Array
Replace Sentrix_ID into Slide
[ Section 1: Read PD file Done ]
[ Section 2: Read IDAT files Start ]
Loading:../../example_datasets/epicv2/mystery_data_2/111111111111/111111111111_R01C01_Grn.idat ---- (1/9)
Loading:../../example_datasets/epicv2/mystery_data_2/111111111111/111111111111_R05C01_Grn.idat ---- (2/9)
Loading:../../example_datasets/epicv2/mystery_data_2/111111111111/111111111111_R07C01_Grn.idat ---- (3/9)
Loading:../../example_datasets/epicv2/mystery_data_2/333333333333/333333333333_R04C01_Grn.idat ---- (4/9)
Loading:../../example_datasets/epicv2/mystery_data_2/333333333333/333333333333_R08C01_Grn.idat ---- (5/9)
Loading:../../example_datasets/epicv2/mystery_data_2/111111111111/111111111111_R03C01_Grn.idat ---- (6/9)
Loading:../../example_datasets/epicv2/mystery_data_2/222222222222/222222222222_R07C01_Grn.idat ---- (7/9)
Loading:../../example_datasets/epicv2/mystery_data_2/222222222222/222222222222_R02C01_Grn.idat ---- (8/9)
Loading:../../example_datasets/epicv2/mystery_data_2/111111111111/111111111111_R04C01_Grn.idat ---- (9/9)
Loading:../../example_datasets/epicv2/mystery_data_2/111111111111/111111111111_R01C01_Red.idat ---- (1/9)
Loading:../../example_datasets/epicv2/mystery_data_2/111111111111/111111111111_R05C01_Red.idat ---- (2/9)
Loading:../../example_datasets/epicv2/mystery_data_2/111111111111/111111111111_R07C01_Red.idat ---- (3/9)
Loading:../../example_datasets/epicv2/mystery_data_2/333333333333/333333333333_R04C01_Red.idat ---- (4/9)
Loading:../../example_datasets/epicv2/mystery_data_2/333333333333/333333333333_R08C01_Red.idat ---- (5/9)
Loading:../../example_datasets/epicv2/mystery_data_2/111111111111/111111111111_R03C01_Red.idat ---- (6/9)
Loading:../../example_datasets/epicv2/mystery_data_2/222222222222/222222222222_R07C01_Red.idat ---- (7/9)
Loading:../../example_datasets/epicv2/mystery_data_2/222222222222/222222222222_R02C01_Red.idat ---- (8/9)
Loading:../../example_datasets/epicv2/mystery_data_2/111111111111/111111111111_R04C01_Red.idat ---- (9/9)
Extract Mean value for Green and Red Channel Success
Your Red Green Channel contains 1105209 probes.
[ Section 2: Read IDAT Files Done ]
[ Section 3: Use Annotation Start ]
Reading EPICv2 Annotation >>
!!! Important, since version 2.29.1, ChAMP set default `EPIC` arraytype as EPIC version 2.
You can set 'EPIC' or 'EPICv2' to use version 2 EPIC annotation
If you want to use the old version (v1), please specify arraytype parameter as `EPICv1`.
For 450K array, still use `450K`
Fetching NEGATIVE ControlProbe.
Totally, there are 411 control probes in Annotation.
Your data set contains 411 control probes.
Generating Meth and UnMeth Matrix
Extracting Meth Matrix...
Totally there are 937055 Meth probes in EPICv2 Annotation.
Your data set contains 937055 Meth probes.
Extracting UnMeth Matrix...
Totally there are 937055 UnMeth probes in EPICv2 Annotation.
Your data set contains 937055 UnMeth probes.
Generating beta Matrix
Generating M Matrix
Generating intensity Matrix
Calculating Detect P value
Counting Beads
[ Section 3: Use Annotation Done ]
[<<<<< ChAMP.IMPORT END >>>>>>]
[===========================]
[You may want to process champ.filter() next.]
[===========================]
[<<<< ChAMP.FILTER START >>>>>]
-----------------------------
In New version ChAMP, champ.filter() function has been set to do filtering on the result of champ.import(). You can use champ.import() + champ.filter() to do Data Loading, or set "method" parameter in champ.load() as "ChAMP" to get the same effect.
This function is provided for user need to do filtering on some beta (or M) matrix, which contained most filtering system in champ.load except beadcount. User need to input beta matrix, pd file themselves. If you want to do filterintg on detP matrix and Bead Count, you also need to input a detected P matrix and Bead Count information.
Note that if you want to filter more data matrix, say beta, M, intensity... please make sure they have exactly the same rownames and colnames.
[ Section 1: Check Input Start ]
You have inputed beta,intensity for Analysis.
pd file provided, checking if it's in accord with Data Matrix...
pd file check success.
Parameter filterDetP is TRUE, checking if detP in accord with Data Matrix...
detP check success.
Parameter filterBeads is TRUE, checking if beadcount in accord with Data Matrix...
beadcount check success.
parameter autoimpute is TRUE. Checking if the conditions are fulfilled...
!!! ProbeCutoff is 0, which means you have no needs to do imputation. autoimpute has been reset FALSE.
Checking Finished :filterDetP,filterBeads,filterMultiHit,filterSNPs,filterNoCG,filterXY would be done on beta,intensity.
You also provided :detP,beadcount .
[ Section 1: Check Input Done ]
[ Section 2: Filtering Start >>
Filtering Detect P value Start
The fraction of failed positions per sample
You may need to delete samples with high proportion of failed probes:
Failed CpG Fraction.
sample1 0.008623827
sample2 0.009623768
sample3 0.010284348
sample4 0.008498968
sample5 0.008403989
sample6 0.010018622
sample7 0.012234074
sample8 0.006075417
sample9 0.008782836
Filtering probes with a detection p-value above 0.01.
Removing 23273 probes.
If a large number of probes have been removed, ChAMP suggests you to identify potentially bad samples
Filtering BeadCount Start
Filtering probes with a beadcount <3 in at least 5% of samples.
Removing 12573 probes
Filtering NoCG Start
Only Keep CpGs, removing 3508 probes from the analysis.
Filtering SNPs Start
!!! Important, since version 2.29.1, ChAMP set default `EPIC` arraytype as EPIC version 2.
You can set 'EPIC' or 'EPICv2' to use version 2 EPIC annotation
If you want to use the old version (v1), please specify arraytype parameter as `EPICv1`.
For 450K array, still use `450K`
Using general mask options
Removing 30917 probes from the analysis.
Filtering MultiHit Start
Filtering probes that align to multiple locations as identified in Nordlund et al
Removing 0 probes from the analysis.
Filtering XY Start
Filtering probes located on X,Y chromosome, removing 20480 probes from the analysis.
Updating PD file
Fixing Outliers Start
Replacing all value smaller/equal to 0 with smallest positive value.
Replacing all value greater/equal to 1 with largest value below 1..
[ Section 2: Filtering Done ]
All filterings are Done, now you have 846304 probes and 9 samples.
[<<<<< ChAMP.FILTER END >>>>>>]
[===========================]
[You may want to process champ.QC() next.]
[<<<<< ChAMP.LOAD END >>>>>>]
[===========================]
[You may want to process champ.QC() next.]
champ.QC(beta = myData_2$beta,
pheno=myData_2$pd$Sample_Group,
resultsDir="./CHAMP_QCimages/")
[===========================]
[<<<<< ChAMP.QC START >>>>>>]
-----------------------------
champ.QC Results will be saved in ./CHAMP_QCimages/
[QC plots will be proceed with 846304 probes and 9 samples.]
<< Prepare Data Over. >>
<< plot mdsPlot Done. >>
<< Plot densityPlot Done. >>
< Dendrogram Plot Feature Selection Method >: No Selection, directly use all CpGs to calculate distance matrix.
<< Plot dendrogram Done. >>
[<<<<<< ChAMP.QC END >>>>>>>]
[===========================]
[You may want to process champ.norm() next.]